




Drug-resistant tuberculosis (TB) remains a major problem of the public health system. In 2022, TB was newly diagnosed and officially notified in ≈7.5 million persons, and the total number of deaths caused by TB was 1.30 million. In 2019, TB was the 13th leading cause of death worldwide and the leading cause of death from a single infectious agent (1). The incidence of drug-resistant TB is estimated to be >400,000 new cases (1).
The limited number of effective TB drugs has pushed the development of new candidates. Bedaquiline, introduced in 2012, began the new era of therapy and was successfully used in combinations with linezolid, clofazimine, and pretomanid/delamanid in new regimens, including all-oral short-course regimens for drug-resistant TB (2).
Bedaquiline has a novel distinct mode of action on bacterial physiology by blocking the ATP synthase of Mycobacterium tuberculosis, thus causing the decrease of the ATP pool (3) and consequent cell death (4). Two main mechanisms of bedaquiline resistance by alteration of the target and drug efflux were identified in vitro (3,5) and, subsequently, in clinical isolates (6–9).
In the first main mechanism of resistance, the iron-scavenger siderophore transporter complex MmpS5/L5 (10) provides bedaquiline efflux from the cytoplasm. Mutations in the repressor gene mmpR5(rv0678) lead to derepression of the operon mmpS5-mmpL5, thus lowering the bedaquiline concentration inside the cell (5). Mutations are spread along the open reading frame and include insertions, deletions, premature stop-codons, and amino acid substitutions (11). The most prevalent type of mutations is nucleotide insertion or deletion in homopolymeric sequences in the gene (12). Drug-resistant isolates with mmpR5 mutations that emerged during bedaquiline treatment have been described in many regions of the world (13–15).
The second type of mutations leading to bedaquiline resistance occur in the c subunit of ATP synthase, encoded by the atpE gene (3). Amino acid substitutions at particular positions in the protein prevent the drug binding (16) and are rarely identified in clinical strains (12). Isolates with atpE mutations possess higher bedaquiline MICs and are supposed to emerge after repressor-inactivating mutations in clinical strains (17).
Other genetic traits have been proposed to cause bedaquiline resistance; however, the number of such cases is low, and the World Health Organization (WHO) has associated substitutions in only atpE, mmpR5, and pepQ genes with resistance to bedaquiline (18). Most of those mutations are in interim status because of insufficient statistics of characterized isolates with a particular mutation. Only the combined category of loss-of-function mutations in the mmpR5 gene and selected frameshifting mutations in hot spots are classified as category 1 mutations. WHO experts indicate that the functionality of the mmpS5 and mmpL5 genes should also be validated because of epistatic interactions with repressor mutations (19). In this study, we analyzed bedaquiline-resistant clinical strains isolated at Novosibirsk TB Research Institute (Novosibirsk, Russia), analyzed mutation profiles using whole-genome sequencing, and proposed the participation of novel determinants of bedaquiline resistance. The study protocol was approved by the Ethical Committee of the Federal State Budgetary Institution Novosibirsk TB Research Institute (protocol no. #52/1, May 5, 2021).
Clinical Isolates and Drug Susceptibility Testing
Clinical isolates were obtained from patients of the Novosibirsk TB Research Institute in whom TB had first been diagnosed during 2006–2018 (Table 1). In the study period (2021–2022), those patients received treatment with a bedaquiline and linezolid–based regimen. Isolates were obtained using liquid media in the Bactec MGIT 960 system (BD, https://www.bd.com) and were further used for drug susceptibility tests, storage, and DNA isolation for molecular tests.
We performed bedaquiline and linezolid susceptibility tests using the modified proportional method in the automatic Bactec MGIT 960 system. Russia’s national guidelines for TB treatment are in accordance with WHO guidelines (20), and we used currently approved critical concentrations for bedaquiline and linezolid of 1 mg/L for both tests. We dissolved and diluted bedaquiline (Molekula Ltd., https://molekula.com) in DMSO and added 100 μL per MGIT tube. We dissolved linezolid (Glenmark Pharmaceuticals, https://glenmarkpharma.com) in sterile H2O as recommended in the guidelines.
Analysis of the CRyPTIC Database
For phylogenetic and mutation frequency analysis, we used a dataset consisting of 9,941 of 12,288 isolates that were sequenced and analyzed by the CRYpTIC Consortium (21). The remaining 2,347 isolates had an ambiguous description of amino acid substitutions and were omitted. We built the phylogenetic tree by nearest-neighbor method in MEGA 11 software (https://www.megasoftware.net) using the pairwise distances calculated from genome mutations. We omitted highly repetitive PE, PPE, PE-PGRS genes, and insertion elements (Appendix Figure). We verified the reliability of the phylogenetic tree by clonal distribution of selected lineage-specific single-nucleotide polymorphisms. We assembled data consisting of the phylogenetic tree, isolate susceptibility profiles, and mutation profiles in a local database, powered by custom Python scripts.
Molecular Determinants of Resistance to Bedaquiline and Linezolid
During June 2021–May 2022, we identified 9 cases of culture positivity long after the initiation of treatment with bedaquiline plus linezolid–based regimens. We performed drug susceptibility tests for bedaquiline and linezolid using the proportion method. Of the 9 isolates, 8 were resistant to bedaquiline; 2 of the 8 bedaquiline-resistant isolates and the 1 bedaquiline-susceptible isolate were resistant to linezolid (Table 1).
In all 9 isolates, including the susceptible isolate, we identified mutations in either mmpR5 or atpE genes (Table 1). Most of the mutations identified were in mixed state with wild-type sequence, so the allele frequencies were estimated by calculating the relative number of reads with mutations. Unexpectedly, 4 of the 8 resistant strains had substitutions in 61 and 66 codons of the atpE gene, which are rarely reported in bedaquiline-resistant isolates. The 100% mutated allele frequency at the atpE locus correlated with the absence of substitutions in the mmpR5 gene in 3 isolates (#5, #7 and #8). Another isolate (#9c) in addition to AtpE Glu61Asp in the mixed state also had the loss-of-function frameshifting MmpR5 Ile67fs and substitution MmpL5 Asn772Thr, which could affect bedaquiline resistance.
In 6 of 9 isolates, we observed variable mutations of mmpR5, predominately leading to frameshifts. Single-nucleotide insertions were located in 2 hot spots: around codons 46–49 (3 cases) and 67 (2 cases). Another type of mutations, the amino acid substitution Ser63Gly in MmpR5, was observed only in 1 case with an allele frequency of ≈75%. Serine 63 is located in the turn part of the HTH DNA binding domain of the MmpR5 repressor (22), providing the rationale for the effect of substitutions at this point on bedaquiline resistance. One isolate susceptible to bedaquiline had an inactivating mutation in the mmpR5 gene with an allele frequency close to 100% and mutation Val165Leu in the atpB gene encoding the a subunit of ATPsynthase and located just upstream of the atpE gene (Table 1). This isolate belongs to the Central Asia/Russian genotype, contrary to Beijing B0/W148 for all other isolates. Resistance to linezolid in 3 isolates was caused by the canonical substitution Cys154Arg in the ribosomal protein RplC (23).
Candidate Markers of Resistance to Bedaquiline
We also sequenced 3 consequent isolates from the same patient, who was treated with a bedaquiline-based regimen. Although they were isolated during only half a year, we observed the dynamic change in mmpR5 mutations and the emergence of AtpE(Glu61Asp) amino acid substitution (Table 2). Thus, in the latest sample, allele frequency of AtpE(Glu61Asp) was 59%, and MmpR5(Ile67fs) was ≈100%. Another mutation MmpR5(Leu143stop) was highly represented in the first sample in the series and then gradually disappeared. Of note, we observed that the additional substitution in the efflux pump subunit MmpL5(Asn772Thr) emerged simultaneously with the AtpE substitution.
Allele frequencies of mutations associated with resistance to other drugs and mutations in genes possibly associated with virulence and host-adaptation also changed in sequential isolates. Furthermore, 3 synonymous amino acid substitutions were recorded in the latest isolate #9c.
In addition to the isolates from case #9, isolates #2 and #4 also had mutations that could be associated with bedaquiline resistance. In those cases, the reference susceptible strains isolated before the start of the treatment were not available, and the emergence of mutations cannot be confirmed directly. The bedaquiline susceptible isolate #4 had Val165Leu substitution in a subunit of ATPase, encoded by the atpB gene, in addition to the loss-of-function MmpR5(Glu49fs) (Table 1). Another bedaquiline-resistant isolate (#2) had a frameshifting mutation in the mmpL4 gene together with a loss-of-function mutation in mmpR5.
Resistance of M. tuberculosis to bedaquiline is driven by 2 main mechanisms, drug efflux by the MmpS5-MmpL5 complex and alteration of its binding site in the rotor part of ATP synthase (3,5). Mutations in both repressor gene mmpR5 and atpE encoding the c subunit emerge during bedaquiline treatment. Mutations in mmpR5 are distributed along the open reading frame, lead to an increase in MIC, and are found more frequently in clinical strains.
We identified a high prevalence of the atpE mutations in clinical isolates from patients who were treated with the bedaquiline-linezolid treatment scheme and remained culture-positive long after the start of the treatment. Most isolates developed phenotypic resistance to bedaquiline detected by Bactec MGIT 960 with a recommended critical concentration of 1 mg/L. Four of 8 bedaquiline-resistant strains had amino acid substitutions Glu61Asp or Ile66Met of the c subunit in positions that are responsible for the direct interaction with bedaquiline. Those substitutions are classified by the WHO as associated with resistance-interim (18).
Only 17 clinical MTB isolates with atpE mutations have been described in 9 studies (9,12,24–30). Another study from China also describes the existence of AtpE Asp28Gly and Ala63Pro substitutions in clinical strains resistant to bedaquiline; however, the number of such isolates cannot be extracted from the published data (31). The most frequent mutation in AtpE was Ile66Met, found in 6 cases. The same dominance was also found in this study: 2 of 4 isolates bore the same substitution. It could be assumed that this mutation provides the best balance between loss of susceptibility to bedaquiline and fitness cost of altered structure of ATP synthase. However, in a report from France, such atpE mutation was transient in a series of clinical isolates from 1 patient and was lost in subsequent isolates; the patient was successfully cured (28).
The effect of particular mutations in mmpR and atpE on the bedaquiline MIC value is variable. Part of mutations in the mmpR5 repressor gene lead to a modest increase in MIC close to the borderline value and thus are considered not associated with resistance (32). Of note, even loss-of-function mutations in the repressor, which are supposed to lead to complete derepression of the efflux operon mmpS5-mmpL5, do not invariantly lead to resistance, as was found for an isolate in this study. In this case, the determination of MIC could have been particularly beneficial to estimate the effect of mutation on phenotype; the proportion method with currently approved critical concentration definitely limits the study.
Other genetic traits could affect the phenotype by epistatic interaction, as was shown for the isolate with both derepression of the efflux operon and the inactivated efflux gene (19). In general, the particular genotype of the pathogen could have an effect not only on transmission dynamics but also on the speed of resistance acquisition and survival under drug-induced stresses (33). In Russia, 2 main genotypes of lineage 2 are associated with resistance to many drugs and rapid transmission in the population, B0/W148 and Central Asia/Russia (34). At least 12 of 17 previously reported atpE mutants belong to either of these sublineages; 10 of them were reported in different regions of Russia. Only 2 strains belong to lineage 4 (24) and lineage 3 (27). All bedaquiline-resistant isolates reported here belong to Beijing B0/W148, whereas the single susceptible isolate belongs to the Beijing Central Asia/Russia sublineage.
The recommended critical concentration of 1 mg/L for resistance determination using the Bactec MGIT 960 is probably too high (32). A substantial overlap of MIC values at 0.125 mg/L and 0.25 mg/L was previously reported for pairs of exposed and nonexposed and wild-type and mutated isolates (12). On the basis of those observations, the critical concentration should be lowered to at least 0.25 mg/L for better differentiation of resistant and susceptible isolates. The current value enables identification of strains with substantially elevated MIC, whereas intermediate resistance is missed by the phenotypic method. Thus, the detection of a considerable number of isolates with AtpE substitutions, which are associated with high bedaquiline MICs, was not unexpected. In addition, we identified strains with mmpR5 mutations only, including both frameshifting and amino acid substitutions such as Ser63Gly, which occurred in the HTH DNA binding domain of the repressor.
Contrary to several previous reports, we did not find pepQ or rv1979c mutations in bedaquiline-resistant isolates (15,35–37). Although the isolated mutations in pepQ are rare and usually are accompanied with mmpR5 mutations, WHO has stated that pepQ mutations are associated with resistance (18). We propose that mmpL5, mmpS4-mmpL4, and atpB genes could also affect the bedaquiline-resistance phenotype from the analysis of microevolution events and the identification of mutations in single isolates in heteroresistant state, which also point to the ongoing evolution of the pathogen.
We observed mutations in genes that encode the siderophore export transport proteins MmpL5/S5 and MmpL4/S4 (10). First, the amino acid substitution Asn772Thr of the efflux MmpL5 emerged in the latest of series of 3 consequent isolates from 1 patient, together with fixation of loss-of-function variant of the mmpR5 gene and the emergence of AtpE Glu61Asp. The mmpR5 mutation has an allele frequency close to 100%, pointing to the fixation of resistance-associated variant. The allele frequency of MmpL5 Asn772Thr was 40% and for AtpE Glu61Asp was 60%, resulting in a total of 100%. Therefore, we could speculate the existence of 2 strains, 1 with the simultaneous presence of MmpR5(Ile67fs) and AtpE(Glu61Asp) and the other with MmpR5(Ile67fs) and MmpL5(Asn772Thr). If substitution in MmpL5 decreases susceptibility to bedaquiline, both strains would survive better during bedaquiline treatment and have higher MICs than the parental mutant mmpR5 strain.
Figure 1
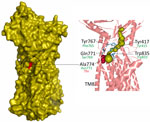
Figure 1. Mapping of the candidate substitution Asn722Thr on the atomic model of Mycobacterium smegmatis MmpL5 transporter (PDB: 9B46) in study of high prevalence of atpE mutations in bedaquiline-resistant …
Recently, the cryo-EM structure of MmpL5 from M. smegmatis was determined, and possible root of transport of mycobactin and other molecules was proposed on the basis of channel prediction, molecular modeling, and docking calculations (38). Of note, Ala774 of the M. smegmatis, which corresponds to Asn772 of M. tuberculosis, is located in the transmembrane domain TM8, responsible for opening and closing of the channel (Figure 1). More precisely, Ala774 is located in the cavity that forms the channel inlet and the narrowest region of the channel together with Tyr767, Gln771, Tyr417, and Trp835, highlighted in the study (38). This model should nevertheless be used with caution, taking into account the MmpS5-guided trimerization of MmpL5 and anchoring of the complex in membrane, which is necessary for bedaquiline efflux (39).
Mutations in the mmpL5 are not frequent; however, MmpL5/S5 efflux does not appear to be indispensable, since a substantial number of frameshift mutations were found in different sublineages of M. tuberculosis (19), and cell wall morphology is not affected in strains with deletion of mmpL5 or mmpS5 (39). According to the WHO, mutation in the same codon was found earlier; MmpL5(Asn772Ser) has uncertain significance in the resistance to bedaquiline and clofazimine (18). Two strains with this mutation in the CRyPTIC database have a low bedaquiline MIC of 0.03 mg/L (21).
More notably, the loss-of-function mutation of mmpL4 was found in a bedaquiline-resistant isolate together with frameshift in the mmpR5. Both the MmpL5/MmpS5 and MmpL4/MmpS4 transporter complexes secrete iron-scavenging siderophores mycobactin and carboxymycobactin (10). Although the role of MmpL5/MmpS5 in bedaquiline and clofazimine resistance is well established as the export pump of the drugs, mutations in mmpS4/L4 were not previously associated with bedaquiline resistance. However, a recent study has shown that deletion of mmpS4 or mmpL4 increases bedaquiline MIC from 2 to 4 times in vitro, probably because of the compensatory upregulation of the mmpS5–mmpL5 operon (40). The 91% allele frequency of the frameshifting variant of mmpL4 found in isolate #2 points to its recent emergence.
To confirm the significance of our findings, we retrospectively analyzed the genomes of clinical isolates of patients treated with bedaquiline from our previous reports (9,12). Indeed, in a series of consequent isolates from a patient who was treated with a bedaquiline-containing regimen, a similar loss-of-function mutation of mmpL4 caused by frameshift in the 341 codon emerged (Table 3). The strain already had a high bedaquiline MIC caused by the presence of amino acid substitutions in AtpE and MmpR5. However, in the 2 latest isolates, the emergence of the mutation in mmpL4 was accompanied by a further increase in bedaquiline MIC, proportional to the allele frequencies, thus confirming the effect of loss-of-function mutations in the mmpL4 gene.
The third gene, which was also supposed to have an effect on bedaquiline resistance, was the atpB gene encoding a subunit of the ATP synthase. This subunit is a part of the stator, lying in tight contact with the rotor, which consists of 9 identical c subunits encoded by the atpE gene. Moreover, in the ATP synthase operon atpB gene is positioned right upstream of atpE, and mutations in bedaquiline resistant isolates were already described (24). However, those mutations were found close to the 3′ end of the gene (codons 222, 244, and 250) and were supposed to alter the transcription of the downstream atpE gene.
Figure 2
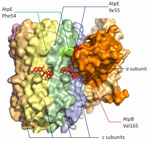
Figure 2. Mapping of the candidate substitution Val165Ile in AtpB on the atomic model of Mycobacterium tuberculosis ATP synthase (PDB: 8J0S) in study of high prevalence of atpEmutations…
We identified the substitution of Val165, which is located at the a/c interface close to the lagging binding site of bedaquiline in ATP synthase (41). It interacts with Phe54 and Ile55 of the c subunit, and substitution for a larger Leucine could change the structure substantially (Figure 2). This strain also had a loss-of-function mutation of mmpR5 and was susceptible to bedaquiline. Therefore, the particular role of AtpB Val165Ile cannot be established from our data.
The CRyPTIC database contains 18 isolates with the neighboring AtpB(Thr166Met) (21). They belong to the same clone inside lineage 3 isolated in different laboratories (Appendix Figure). They do not contain mutations in genes associated or involved in bedaquiline resistance: atpE, mmpR5, pepQ, mmpL5, mmpS5, mmpL4, and mmpS4. Furthermore, no other isolates with other mutations in atpB were found in the CRyPTIC study. The MIC values of those isolates were slightly shifted toward higher values compared with the entire set of strains, pointing to the possible involvement of this substitution in decreased susceptibility to bedaquiline (Table 4).
The frequencies of mutations, associated with resistance to other drugs, also changed in the series obtained from 1 patient. Therefore, the substitution in the alr gene encoding alanine racemase (42,43), associated with resistance to D-cycloserine, has disappeared in the third isolate #9c, which probably points to its high fitness cost. However, another mutation in the gabD2 gene, previously associated with resistance to D-cycloserine (44), emerged. The lineage 4 strains with the loss-of-function gabD2 mutation were more frequently identified in patients in the epidemiologic study from Colombia (45), thus associated with success in transmission, which could be affected also by reduced susceptibility to drugs.
Mutated variants of 2 genes, rv2690c and ceoC, previously associated with resistance to pyrazinamide (for rv2690c) (46) and isoniazid (for ceoC) (47), emerged in the latest isolate. Those genes could be responsible for virulence and host adaptation also (48,49). Other genes associated with virulence, such as rv2298 and 2 PPE genes, also changed the frequencies of mutated alleles in sequential isolates.
Research of the microevolution of the pathogen within the host has allowed for clarity into genetic traits related to resistance and host adaptation, thus complementing the massive body of genome-wide association studies. However, it is hard to discern whether the mutations emerged internally, under selective pressures from treatment or the immune system of the host, or are neutral, from genetic drift caused by spread in the general population. In addition, the exact pressure is not known because of poor adherence to treatment in most chronic cases and individual variability in pK/pD (50).