




[ad_1]
Disclaimer: Early release articles are not considered as final versions. Any changes will be reflected in the online version in the month the article is officially released.
Author affiliation: Statens Serum Institut, Copenhagen, Denmark (A. Gewecke, R.N. Sieber, S. Hallstrøm, M. Stegger, M.C. Arendrup); Harry Butler Institute, Murdoch University, Perth, Western Australia, Australia (M. Stegger); dansk MiljøAnalyse, Vedbæk, Denmark (B. Kolarik); Copenhagen University Hospital, Rigshospitalet, Copenhagen (J.D. Knudsen, A. Hall, N.H. Vissing, M.C. Arendrup); Aalborg University, Copenhagen (B. Andersen); University of Copenhagen, Copenhagen (M.C. Arendrup)
Aspergillus flavus, the second leading cause of invasive aspergillosis (IA) worldwide, is an opportunistic mold that can cause severe infection, including IA, in immunocompromised patients (1). It predominates in Asia, the Middle East, and Africa because of its resilience in arid climates; however, because of climate change, its presence in the Northern hemisphere is expected to increase (1; N. van Rhijn, unpub. data, https://www.researchsquare.com/article/rs-6545782/v1). Hospital outbreaks mainly occur when airborne conidia are dispersed during construction work (2). The lower respiratory tract is usually the affected site of infection; in hematology patients, fatality rates are often high (2).
Renovations during 2017–2019 in a pediatric hematology ward at the largest tertiary hospital in Denmark likely led to infections in 6 patients by spreading conidia (3). Infections occurred despite amphotericin B prophylaxis, consistent with the intrinsic reduced susceptibility of A. flavus. Transition to posaconazole prophylaxis was implemented and prevented A. flavus in patients with adequate serum drug levels. In 2022–2023, we examined the genetic epidemiology of the outbreak using short tandem repeat (STR) genotyping, STRAfla (4,5). We confirmed isogeneity and linked 12 additional patients to the outbreak. Analysis of air samples also indicated the presence of A. flavus outbreak isolates in the ward, suggesting its undetected perseverance in the hospital over time. However, 8 more patients positive for outbreak-related A. flavus appeared during April 2023–January 2024; of those, 3 were from the outbreak ward floor and 5 were from other parts of the hospital, including a staff member in the Department of Clinical Microbiology (DCM).
In early 2024, a multidisciplinary team of specialists was formed to control the outbreak. In this study, we aimed to resolve the outbreak through optimizing environmental sampling to guide cleaning and source investigation and by using genome sequencing on a broad panel of A. flavus from the outbreak, the hospital environment, and epidemiologically unrelated patients and sites. We also used time-scaled phylogeny to understand the evolution of the outbreak isolates. Although the comparison of typing methodologies is not a novel approach in a clinical context (6–10), this study also applied an extensive genomics approach with time-scaled phylogeny to an ongoing Aspergillus outbreak. Leveraging phylogenetics delineated the evolution of isolates within an A. flavus population that inhabited the hospital environment and successfully pinpointed a plausible source of contamination, as well as an unexpected timing of introduction.
Environmental Sampling and Comparison of Sampling Strategies
We took air samples (1 m3) using either a Microbio MB1 bioaerosol sampler (Cantium Scientific, https://www.cantiumscientific.com) onto 5 cm-diameter V8 agar contact plates (Bioneer A/S, https://bioneer.dk) or a MBASS30 V3 air sampler (Holbach, https://www.holbach.biz) onto 9-cm diameter V8 agar petri dishes (Bioneer A/S). We took surface samples using 5-cm diameter (25 cm2) V8 agar contact plates (Bioneer A/S), preferably from infrequently cleaned surfaces (11). We incubated 1 set of contact plates and petri dishes at 25°C and read after 7 days and incubated the other set at 37°C and read after 3 days. Air sampling was nonaggressive because disturbing dust was unacceptable for hospital staff.
Figure 1
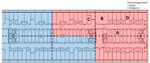
Figure 1. Floorplan of the outbreak floor in hospital South complex in study of environmental and phylogenetic investigations of Aspergillus flavusoutbreak linked to contaminated building materials, Denmark, 2025. The hospital…
To assess A. flavus contamination and general fungal load, hospital sampling in March–June 2024 yielded 449 samples from 299 locations (in total). In March, we conducted environmental sampling at 41 predefined locations using V8 agar (12). At each location, we collected 4 samples (164 samples in total): 1 air sample incubated at 25 °C, 1 air sample incubated at 37 °C, 1 surface sample incubated at 25 °C, and 1 surface sample incubated at 37 °C. In addition, we investigated outdoor air controls and some sporadic surface samples (24 samples from 12 locations). Thus, in total, we collected 188 samples from 53 locations in the pediatric outbreak ward and outside the hospital. In April–June, we conducted another session focused on surface sampling and culturing at 37°C from the outbreak floor and collected 97 samples from 87 locations in both the pediatric and the adult hematology ward (Figure 1). That secondary sampling focused on areas that were most contaminated with A. flavus in the initial (paired) sampling session. Outside the outbreak floor, we took another 164 samples from 159 locations in high activity areas. We compared A. flavus CFU counts for paired samples in Rstudio using a χ2 test (using table 2×2 function, R version 4.2.3, and the package Publish) (The R Project for Statistical Computing, https://www.r-project.org).
We verified active growth of A. flavus on materials using tape preparations, microscopy (12), and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for identification (13). We collected soil samples (n = 8) from the hospital surroundings near the outbreak-affected South complex using a method described elsewhere (14) and cultured them at 37°C. Four A. flavus isolates were previously isolated from wood-based building materials, purchased from 2 retailers in Denmark, in an unrelated study of precontaminated materials (B. Andersen, unpub. data).
STRAfla of Environmental Isolates
We applied STRAfla genotyping on 145 A. flavus isolates as previously described (4,5). Of those, 33 isolates (24 from patients and 9 from air) were included in our previous study (5). We genotyped additional patient isolates (n = 11), hospital interior isolates (n = 87 [1 isolate per positive location]), soil isolates (n = 10), and building material isolates (n = 4).
Whole-Genome Sequencing of A. flavus Isolates
The dataset included 92 clinical and 17 environmental isolates from the outbreak hospital, 4 from unrelated wood-based materials, and 54 comparator isolates (167 isolates in total). All comparators, except 2, were previously STRAfla typed (5), and all originated either from hospital patients in Denmark who were not related to the outbreak or were control strains (4 UK-NEQAS and 2 ATCC 204304/200026 [CBS 128202] isolates).
We prepared sequencing libraries using a custom modified version of the DNA prep kit (Illumina, https://www.illumina.com) based on the Hackflex protocol (15) using 20 ng (2 ng/µL) genomic DNA as input and 25 µL of a 2× laboratory-made tagmentation buffer (20 mM Tris-HCl at pH 7.6), 20 mM MgCl2, and 10% (vol/vol) Propylene Carbonate (all Sigma-Aldrich, https://www.sigmaaldrich.com). We indexed the generated libraries with IDT for Illumina Unique Dual indexes and quantified them using the Quant-iT dsDNA HS Assay Kit (Thermo Fisher Scientific, https://www.thermofisher.com). We calculated molar concentrations using a standard conversion factor of 2 and pooled the samples at equimolar ratios. We assessed the quality of the final sequencing libraries by measuring concentration on a Qubit 3.0 fluorometer using the Qubit dsDNA HS quantification assay kit (Thermo Fisher Scientific) and by performing a fragment analysis using a TapeStation 4200 with the D5000 Screen Tape assay kit (Agilent, https://www.agilent.com). We sequenced quality checked final libraries on a NovaSeq (Illumina) using an S4 flowcell and 200-cycle reagent kit generating 2 × 100-bp paired-end reads.
We assessed quality and depth using NASP version 1.2.1 (16) and contamination screened using Kraken version 1.0 (17). We performed assembly with SPAdes version 3.11.1 (18) using the “–isolate” flag. We called single-nucleotide polymorphisms (SNPs) with NASP version 1.2.1 (16) aligned to NRRL 3357 (reference genome CBS128202, GenBank accession no. GCF_014117465) using BWA-MEM (H. Li, unpub. data, https://arxiv.org/abs/1303.3997v2) and GATK, requiring >10× depth and >90% unambiguous base calls. We removed recombination-prone regions using Gubbins version 2.3.4 (19) and inferred phylogenies with IQTREE version 2.0.3 (20) using the integrated model tester with ascertainment bias correction (-m TEST+ASC). Time-scaled phylogeny of cluster 10 used core-genome SNPs with strain T21212 as reference. We used BEAST2 (https://www.beast2.org) with sample dates as tips, BEAST Model Test, a relaxed clock model, and coalescent exponential population, with Markov chain Monte Carlo lengths of 200 million. We inferred clusters using TreeCluster (21) with the Max Clade method at a threshold of 0.01 for the full dataset and 0.001 for the cluster 10 tree. Furthermore, we inferred STRAfla locus 2A ancestral state in R version 4.4.2 using the ace function from the package ape under an equal-rates model.
Environmental Sampling
Figure 2

Figure 2. Distribution of Aspergillus flavus in hospital pediatric hemotology ward in study of environmental and phylogenetic investigations of A. flavusoutbreak linked to contaminated building materials, Denmark, 2025….
For initial sampling and comparison of strategies, paired sampling with culture on V8 agar yielded 1,712 fungal CFU, of which 775 were classical opportunistic species (including Aspergillus flavus, A. fumigatus, A. niger, and Mucorales spp.) and 593 were A. flavus specifically (Table 1; Figure 2). Surface sampling was more sensitive than air sampling overall (1,165 CFU [surface] vs. 547 CFU [air]), for the classical opportunistic species (731 CFU [surface] vs. 30 CFU [air]), and for A. flavus specifically (585 CFU [surface] vs. 8 CFU [air]) in the paired 81 air versus 81 surface samples. Culturing surface samples at 37°C increased A. flavus yield 1.77-fold (95% CI 1.57–1.99-fold; pA. calidoustus, Cladosporium, and Penicillium spp. isolates when incubating at 37°C. Outdoor air controls grew Cladosporium and Penicillium spp. isolates but not A. flavus isolates.
Figure 3

Figure 3. Examples of locations with dust accumulation above ceiling and large Aspergillus flavus contamination from hospital pediatric hemotology ward in study of environmental and phylogenetic investigations of …
We performed subsequent sampling during April–June 2024 with incubation at 37°C in 15 pediatric ward locations, which yielded a total of 324 A. flavus CFU (median 13.0 [range 0–58] CFU/25 cm2). Additional sampling above the ceiling gave 195 A. flavus CFU (median 4.0 [range 0–55] CFU/25 cm2). Samples beneath insulation sheets on the ceiling tiles were negative (Figure 3). In the adjacent adult hematology ward (Figure 1), we found 527 A. flavus CFU (median 1.0 [range 0–53] CFU/25 cm2). On 10 other floors, we collected 164 surface samples; we found A. flavus in 5 samples (33 CFU total); 29 were from a kitchen refrigerator top (central complex) and 4 were scattered elsewhere.
Figure 4

Figure 4. Testing materials from room A in hospital pediatric hemotology ward in study of environmental and phylogenetic investigations of A. flavusoutbreak linked to contaminated building materials, Denmark, 2025. A)…
Peak A. flavus contamination was found in a combined staff meeting and lunchroom (room A), the main ward kitchen (room B), and staff offices (rooms C–D) (Figures 1–3), especially on less frequently cleaned surfaces. In November 2024, directed air samples suggested that the kitchenette base under the sink in room A was a source of contamination; yield was 1,500 CFU/m3. After dismantling, microscopy confirmed growth of A. flavus on a plywood cupboard (Figure 4).
STRAfla of Environmental Isolates
We genotyped a total of 145 A. flavus isolates using STRAfla, revealing 28 unique genotypes (Table 2). The dominant outbreak genotype, A, was identified in 19 isolates from 11 patients and 82/96 indoor hospital isolates and occurred exclusively on the outbreak floor, in both the adult and pediatric hematology wards (Table 2; Figure 1). In the adult ward specifically (Figure 1), 36/38 isolates shared genotype A, whereas 2 were different (Table 2). Eight isolates from 7 patients, and 5 isolates from a contaminated DCM incubator shared genotype B, in which 1 of 9 markers (marker 2A) differed from that of genotype A by 1 repeat. Eight additional patient isolates shared 7–8 markers; genotype A displayed different marker variations across the STRAfla panel (Table 2). Among the 82 hospital interior isolates with genotype A, 2 were from the water-damaged plywood cupboard in room A (Table 2; Figure 4). STRAfla of 4 outbreak-unrelated A. flavus isolates from wood-based panels (medium-density fiberboard, oriented strand board, and plywood) obtained from 2 retailers in Denmark and cultured outside this study, revealed partial similarity to genotype A with 5–8 of 9 shared markers (genotypes C, D, E) (Table 2). Remaining genotypes differed from the outbreak genotype A by >5 markers, as did soil isolates (Table 2).
Whole-Genome Sequencing–Based Population Analysis of A. flavus Isolates
Figure 5

Figure 5. Rooted maximum-likelihood phylogeny of 167 A. flavus isolates (cropped) from study of environmental and phylogenetic investigations of A. flavusoutbreak linked to contaminated building materials, Denmark,…
A total of 167 isolates underwent whole-genome sequencing. We aligned reads to the reference genome and identified core-genome SNPs. After excluding recombination regions, we retained 303,560 SNPs in the reference chromosome for phylogenetic inference. This process produced a structured tree with distinct clusters separated by thousands of SNPs (Figure 5). Outbreak isolates grouped into cluster 10, which distinctly separated from others and showed minimal within-cluster variation indicating high genetic similarity. All cluster 10 isolates originated from the hospital except for 4 building material isolates.
Figure 6

Figure 6. Time-scaled core-genome SNP-based phylogeny of cluster 10, which contained all outbreak isolates, in study of environmental and phylogenetic investigations of A. flavusoutbreak linked to contaminated building materials, Denmark,…
Using an internal outbreak draft genome as reference, resolution increased and enabled further analyses. In the cluster-specific tree (Figure 6), we retained 723 SNPs for time-scaled phylogenetic reconstruction. South complex and building material isolates were close, but the building material isolates clustered more basal alongside 2 isolates from patient 11 (infectious disease clinic). The last common ancestor of South complex and building material isolates likely dates to 2002 (95% highest posterior density [HPD] 1995–2008). South complex expansion occurred around 2017 (95% HPD 2015–2018). Isolates were distinct from the DCM isolates, which expanded around 2020 (95% HPD 2018–2021). A common ancestor to that monophyletic cluster dates back to ≈1990 (95% HPD 1975–2004).
Infectious disease clinic isolates were mainly from outpatients and considered noninfected. Those isolates displayed different STRAfla variants (Figure 6). Patients 6 and 11 were colonized and culture-positive twice. In both cases, the patients retained their original genotypes over a 1-year period. Isolates from patients 5, 8, 9, and 12–14 were regarded as plate contaminants because of growth outside the inoculation zone, lack of clinical signs, or both. European Organisation for Research and Treatment of Cancer classification (into proven, probable, possible, or no infection) remains unverified (22).
Phylogenetic analysis also showed that STRAfla marker 2A was able to consistently differentiate South complex from DCM isolates (26 repeats for South complex vs. 25 repeats for DCM isolates) (Table 2). The ancestral genotype of both were most likely genotype A with 26 repeats on the basis of an inferred ancestral state.
This study highlights some of the challenges faced in an Aspergillus outbreak investigation, the importance of optimal sampling and identification strategies, and the valuable insights provided by modern typing and whole-genome sequencing techniques. Environmental sampling was challenging. Air sampling notably underestimated the overall burden of A. flavus on the outbreak floor, whereas in this investigation surface sampling proved notably more sensitive, not only for A. flavus but also for A. fumigatus and A. niger. Surface sampling established a contamination gradient across the outbreak floor (Figures 2, 3) and thus enabled the discovery of a plywood cupboard with microscopy-confirmed A. flavus growth (room A) (Figure 4). Phylogeny revealed a close link between the outbreak isolates and 4 A. flavus isolates from unused wood-based building materials sourced from 4 retailers in Denmark (Figure 6). Alongside confirmed in situ growth on 1 such material inside the ward (Figure 4), those findings suggest precontaminated building materials as a potential origin of the outbreak. On the basis of 95% HPD interval, a common ancestor to cluster 10 might have entered during initial hospital construction (≈1970s) or during later renovations. As previously speculated, the South complex isolates likely spread during ward renovations during 2017–2019 (5), coinciding with the outbreak onset, in accordance with this study’s detection of clonal expansion in the South complex occurring at that time (Figure 6).
Consensus guidelines regarding how to perform and interpret Aspergillus hospital outbreak investigations are currently sparse and linked to air sampling. Ruiz-Camps et al. (23) recommended a limit of 0.5 fungal CFU/m3 in HEPA-protected air and 25 CFU/m3 in unprotected air but did not specify a culture temperature. Chang et al. (24) suggested limits of Aspergillus CFU/m3 in protective isolation areas and Aspergillus CFU/m3 in HEPA-filtered environments, along with a threshold of 15 fungal CFU/m3 for gross colony counts. Moreover, culture at 37°C was recommended to favor growth of human pathogenic fungi (24). However, Morris et al. (25), using the same thresholds, advised culturing at 28°C. Those discrepancies are not trivial and may result in divergent risk assessment. Optimal growth conditions for A. flavus are 32°C–33°C, substrate water activity of 0.95, and pH 4–6.5 (26,27). However, it grows well at 37°C (28) and can grow at 20°C–40°C, substrate water activity of >0.80, and pH 4–9 (26,27). In addition, A. flavus can grow in a variety of substrates because of its abundant enzyme production (11). In our study, mean total fungal CFU counts varied 9-fold for paired air samples cultured at the 2 temperatures, mainly because of A. calidoustus, Cladosporium, and Penicillium, which dominated spore counts at 25°C but rarely cause infections. Perhaps even more concerning, air sampling was overall notably insensitive to detect A. flavus. Thus, surface sampling from infrequently cleaned sites is key for detection; this sampling substantially increased the yield of A. flavus (as well as of A. fumigatus and A. niger), in agreement with previous observations (11,29). Moreover, this study suggests incubation at 37°C to favor A. flavus (and other opportunistic molds) and to minimize overgrowth of irrelevant species, thereby avoiding difficulties in plate reading and underestimation of the A. flavus burden (Table 1). Finally, the dispersal of spores throughout the outbreak floor indicates that settled dust in less frequently cleaned areas might serve as satellite sources of spores with dust-mediated spread of A. flavus (30).
STRAfla typing identified the outbreak genotype A exclusively on the outbreak floor (Table 2) during the environmental sampling session March–June 2024. Closely related isolates came from 7 additional patients, a DCM incubator (5 isolates) (shared 8/9 markers, genotype B), and from wood-based materials (4 isolates) that shared 5–8/9 (genotypes C, D, E) STRAfla markers with the outbreak genotype A. Of note, individual STRAf markers in A. fumigatus might gain single-repeat variants over serial propagation in vivo and in vitro as a result of microevolution (31); thus, we initially regarded genotype A and B as identical and equally involved in the outbreak despite a 1-repeat difference (Table 2). However, genome-based phylogenetic analysis confirmed that degrees of isogeneity among A. flavus, observed through STRAfla typing were consistent. Single-repeat differences at 2A are likely to be genetically meaningful, and provided laboratories continuously use matching protocol parameters, interpretability in regards to single-repeat deviances should not be a concern (32). The relatively small variations in repeat counts in the genotypes of wood-based isolates might also reflect the evolutionary biology of STR sequences that possibly evolve through small, stepwise repeat changes over time (33,34). Taken together, our results support STRAfla’s continued value as a sensitive and specific frontline screening tool for clinical outbreak investigations. Furthermore, those findings are consistent with those of other studies validating microsatellite assays in fungi through genome sequencing (7,9,10), although species-dependent variation in discriminatory power might exist.
Core-genome phylogeny revealed clusters across the isolate collection; cluster 10 (Figures 5, 6) distinctly separated from other clusters. Cluster 10 isolates only included isolates from the outbreak hospital, except those from building materials, supporting a localized outbreak. Minimal genetic diversity within cluster 10 indicated recent clonal expansion.
Bayesian time-scaled phylogeny identified 3 cluster 10 genetic variants: 1 in the South complex, 1 in external building materials (and patient 11 [Figure 6]), and 1 in the DCM. The variants were detected at different time points, but a common ancestor could date back to the 1970s, coinciding with the construction of the hospital (Figure 6).
Confirmed growth of the South complex variant on a plywood base in the outbreak ward suggests a link to precontaminated wood-based materials, also observed for other molds (35). The plywood was part of a kitchen installed in 2012, raising 2 possibilities: that periodic hospital renovations and occasional water damage promoted growth of already prevalent spores that persisted and evolved within the hospital environment, or that repeated introductions to the hospital occurred, possibly through precontaminated wood-based materials. The second is supported by almost genetically identical A. flavus isolates found in hospital-unrelated wood-based materials from retailers in Denmark, but that theory does not explain the finding of an outbreak isolate in a patient sample as early as 2008. If precontaminated materials were the original source, dormant A. flavus spores might have germinated after water damage (36). Wood-based materials can act as Aspergillus substrates (11), and strains phylogenetically related to the outbreak isolates grew on both plywood and oriented strand board (Table 2; Figure 4). Whether cluster 10 represents a genetically distinct subdivision of A. flavus or whether genome rearrangements (37), other alterations (38), or selective pressures (7,38) drove its hospital expansion remains unresolved. However, our findings raise concern related to the use of wood-based materials (and organic building materials in general) in hospital areas housing immunocompromised patients.
In conclusion, this study underscores the importance of comprehensive and correct environmental sampling and genomic analysis in A. flavus outbreaks. Our understanding of this outbreak was substantially enhanced by the use of phylogenetic analysis. Dormant spores of A. flavus had begun actively growing within the hospital environment, with settled dust acting as a reservoir for conidia. Phylogeny validated STRAfla typing as a sensitive and specific tool for frontline outbreak investigation. Crucially, time-scaled phylogeny linked the outbreak isolates to A. flavus isolated from colonized wood-based materials, implicating the hospital’s original construction or subsequent renovations as the likely source of introduction. Our findings should be taken into consideration in planning for the material composition of future hospitals.
Dr. Gewecke is a PhD student in the Unit of Mycology at Statens Serum Institut, Copenhagen, Denmark. Under the supervision of M.C.A., he is researching Aspergillus outbreaks, including an A. flavus outbreak in a Copenhagen tertiary hospital, as part of his thesis.
We thank Désiré Mageme Nahimana for substantial laboratory assistance.
Approval to conduct this study was granted by the Department for Data Protection and Information Security (DBIS) at Statens Serum Institut, J.nr. 22/01885, after institutional review. Publication permission of the data included in this article was granted by the Danish Data Protection Agency (case no. 2025-52-0173, doc. no. 687861). Both case documents will be provided upon request.
Fungal genome sequences generated in this study are available from the European Nucleotide Archive through the accession numbers provided in Appendix 2. Versions of Figures 5 and 6 were presented on a poster during the 12th Congress on Trends in Medical Mycology, September 19–22, 2025, Bilbao, Spain.
No funding was received for this study. M.C.A. has, over the past 5 years, received research grants/contract work (paid to the SSI) from Cidara, F2G, Gilead, and Scynexis, and speaker honoraria from Astellas, Chiesi, Gilead, and F2G. She is the current chairman of EUCAST-AFST.
AI (ChatGPT5) was used for grammatical improvement of the text.
[ad_2]
Source link